
COMP 204 – Assignment #2
Transcription factors代写 NO LATE ASSIGNMENTS WILL BE ACCEPTED,TO ALLOW US TO DISTRIBUTE SOLUTIONS BEFORE THE MIDTERM EXAM.Submit one Python program
Due date: October 15, 23:59
NO LATE ASSIGNMENTS WILL BE ACCEPTED,
TO ALLOW US TO DISTRIBUTE SOLUTIONS BEFORE THE MIDTERM EXAM.Transcription factors代写
- Submit one Python program on MyCourses, which should contain all your functions.
- Write your name and student ID at the top of theprogram
- For each question, complete the relevant function. Do not change its name or arguments.
- Your program should not print anything. Each function should justreturn the appropriate object.
- Do not use any modules except the math module, or any pieces of codeyou did not write yourself.Transcription factors代写
- For each question, your function will be automatically tested on a varietyof test cases, which will account for 75% of the mark.
- For each question, 25% of the mark will be assigned by the TA based on (i) your appropriate naming of variables; (ii) commenting of your program; (iii) simplicity of your program (simpler =better)
Background information:Transcription factors代写
Transcription factors (TF) are proteins that recognize and bind to DNA, and in doing so regulate the expression of a nearby gene. Each type of TF has an affinity for a particular short DNA pattern. For example, the E-‐box TF likes to bind the sequence CAGCTG, whereas the Runx TF likes to bind CCACA. However, every transcription factor has a certain flexibility in the DNA sequences it can bind to. For example, the E-‐box TF will sometimes bind CAGGTG, or CAGCAG. The Runx TF may also bind CCAGA or ACACA. Biologists can identify DNA sequences bound by a given transcription factor using experiments such as chromatin immunoprecipitation followed by sequence (ChIP-‐seq). The outcome of such an experiment is a set of DNA sequences from the genome that are bound by the TF. For example, a ChIP-‐seq experiment on transcription factor E2F1 may produce the following set of 10 sequences:
ACGATG
ACAATG
ACGATC
ACGATCTranscription factors代写
TCGATC
TCGAGC
TAGATC
TAAATC
AAAATC
ACGATA
These DNA sequences are probably only a small subset of all the sites bound by E2F1 in the genome.Transcription factors代写
We would like to be able to learn a model of what type of pattern E2F1 likes to bind, in order to predict, in the human genome, sites that may have been missed by the experiment. In order to do so, bioinformaticians have developed a model called Position Weight Matrix (PWM). Here, you will learn how this model works, and you will have to implement it in Python.
To build a PWM, one first needs to build a Position Frequency Matrix (PFM). A PFM is matrix (table) of 4 rows and L columns, where L is the length of the binding site sequences. In the E2F1 example, L=6. The entry in row i and column j of the PFM, which we denote PFM[i][j], is the frequency of nucleotide i at position j of the binding sites. For E2F1, based on the 10 sites listed above, the PFM is
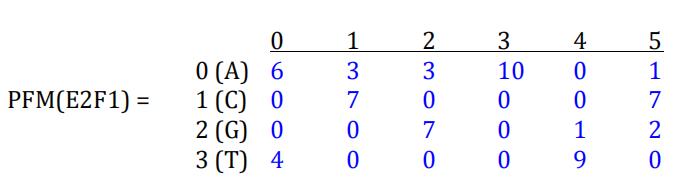
The PFM can be converted to a PWM using the following formula:
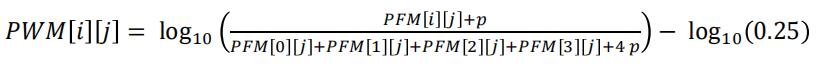
The variable p, called the pseudocount is generally set to a small value such as 0.1. For our E2F1 example, the PWM is:
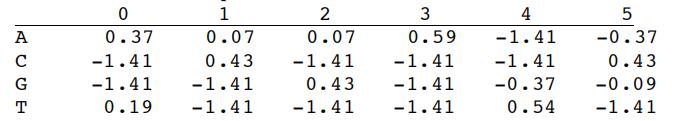
You will notice that large positive values in the PWM (e.g. 0.59) correspond to positions where the TF has a strong preference for a particular nucleotide (e.g. an A at position 3).Transcription factors代写
We can now use a PWM to calculate the score of a match between a candidate sequence of L nucleotides and the PWM. For example, the score of candidate sequence TCGATC is obtained by summing up the appropriate values from the PWM:
Score(TCGATG) = PWM[T][0] + PWM[C][1] + PWM[G][2] + PWM[A][3] + PWM[T][4] + PWM[G][5]
= 0.19 + 0.43 + 0.43 + 0.59 + 0.54 + (-‐0.09) = 2.11
whereas for sequence ACATAG, we get
Score(ACATAG) = PWM[A][0] + PWM[C][1] + PWM[A][2] + PWM[T][3] + PWM[A][4] + PWM[G][5]
= 0.37 + 0.43 + 0.07 + (-‐1.41) + (-‐1.41) + (-‐0.09) = -‐2.03
The larger the score, the higher the affinity of the transcription factor for the sequence.Transcription factors代写
Finally, we can use a PWM to scan a longer sequence to identify potential binding sites for the TF. This is done by calculating the PWM score for every possible portion of L nucleotides of the sequence. For example:
Sequence = ATCGATGAGACTGA Score(0) = Score(ATCGAT) = -‐3.87… Score(1) = Score(TCGATG) = 1.65… Score(2) = Score(CGATGA) = -‐4.16… Score(3) = Score(GATGAG) = -‐4.16… Score(4) = Score(ATGAGA) = -‐0.01… Score(5) = Score(TGAGAC) = -‐2.55…
…
Positions whose score exceeds a user-‐chosen threshold are reported as a putative binding sites for the TF. For example, if we choose a threshold of -‐0.2, then positions 1 and 4 would be reported as putative sites.
Download TFBSscanning.py from MyCourses.
Your work will consist in completing the each of the functions given in this program.
You must not change the function names or their arguments.Transcription factors代写
For each question, it is your responsibility to test your program to make sure it works on all possible cases, not just those provided as examples.

Question 1 (10 points). Encoding of DNA nucleotides into integers.Transcription factors代写
It is often convenient to represent a nucleotide (“A”,”C”,”G”, or “T”) as a number between 0 and 3, where “A” is represented as 0, “C” as 1, “G” as 2, and “T” as 3.
Complete the encode function that takes as argument a string of one character, and returns an integer between 0 and 3. If the string provided as argument is not one of “A”,”C”,”G”, or “T”, the function should return -‐1.
Example of execution: code = encode(“G”) print(code) èè 2
code = encode(“A”) print(code) èè 0 code = encode(“X”) print(code) èè -‐1
Question 2 (20 points). Building a PFM.Transcription factors代写
Write a function called buildPFM that takes as input a list of DNA sequences identified as binding sites for a given transcription factor, and returns a Position Frequency Matrix (PFM), represented as a list of lists of integers, where PFM[nuc][pos] = number of sequences that have nucleotide nuc at position pos. The function can assume that the input sequences all have the same length, but that length is not necessarily 6 (like in our example).
Example of execution:
sites = [“ACGATG”,”ACAATG”,”ACGATC”,”ACGATC”,”TCGATC”,
“TCGAGC”,”TAGATC”,”TAAATC”,”AAAATC”,”ACGATA”]
PFM = buildPFM(sites)Transcription factors代写
print(PFM) èè [[6, 3, 3, 10, 0, 1], [0, 7, 0, 0, 0, 7], [0, 0, 7, 0, 1, 2], [4, 0, 0, 0, 9, 0]]
Note: In your code, you may find it useful to be able to create a PFM with all entries initialized to zero. If L is the length of the desired PFM, this can be done as follows:
PFM = [[0 for i in range(L)] for j in range(4)]
Question 3 (10 points). Building a PWM from a PFM Transcription factors代写
Write the function getPWMfromPFM, which takes as argument a PFM and a real number called the pseudocount and returns a PWM, according to the formula given above.
Example of execution:
PFM = [[6, 3, 3, 10, 0, 1], [0, 7, 0, 0, 0, 7], [0, 0, 7, 0, 1, 2], [4, 0, 0, 0, 9, 0]] PWM = getPWMfromPFM(PFM, 0.1)
print(PWM)
èè Output:
[[0.370356487039949, 0.07638834586345467, 0.07638834586345467, 0.5893480258118245, -‐1.4149733479708178, -‐0.37358066281259295], [-‐1.4149733479708178, 0.43628500074825727, -‐Transcription factors代写
1.4149733479708178, -‐1.4149733479708178, -‐1.4149733479708178, 0.43628500074825727], [-‐
1.4149733479708178, -‐1.4149733479708178, 0.43628500074825727, -‐1.4149733479708178, -‐
0.37358066281259295, -‐0.09275405323689867], [0.19781050874891748, -‐1.4149733479708178,
-‐1.4149733479708178, -‐1.4149733479708178, 0.5440680443502756, -‐1.4149733479708178]]
Question 4 (20 points). Scoring a sequence of length LTranscription factors代写
Write a function score that takes as argument a sequence and a PWM (both of the same length L), and calculates the score of the sequence with that PWM.
Example of execution:
PWM=[[0.370356487039949, 0.07638834586345467, 0.07638834586345467, 0.5893480258118245, -‐1.4149733479708178, -‐0.37358066281259295], [-‐1.4149733479708178,
0.43628500074825727, -‐1.4149733479708178, -‐1.4149733479708178, -‐1.4149733479708178,Transcription factors代写
0.43628500074825727], [-‐1.4149733479708178, -‐1.4149733479708178, 0.43628500074825727,
-‐1.4149733479708178, -‐0.37358066281259295, -‐0.09275405323689867], [0.19781050874891748, -‐1.4149733479708178, -‐1.4149733479708178, -‐1.4149733479708178, 0.5440680443502756, -‐1.4149733479708178]]
s= score(“TCGATG”,PWM) print(s) èè 2.1110425271706337
s=score(“ACATAG”,PWM) print(s) èè -‐2.039670915526873
Question 5 (20 points). Identifying matches in longer sequences
Write a function predictSites that takes as input a DNA sequence and a PWM as positional arguments, as well a floating point number called threshold as keyword argument, and returns a list of the starting positions of substrings whose score with the given PWM is larger or equal to the threshold.Transcription factors代写
Example of execution:
PWM=[[0.370356487039949, 0.07638834586345467, 0.07638834586345467, 0.5893480258118245, -‐1.4149733479708178, -‐0.37358066281259295], [-‐1.4149733479708178,
0.43628500074825727, -‐1.4149733479708178, -‐1.4149733479708178, -‐1.4149733479708178,
0.43628500074825727], [-‐1.4149733479708178, -‐1.4149733479708178, 0.43628500074825727,Transcription factors代写
-‐1.4149733479708178, -‐0.37358066281259295, -‐0.09275405323689867], [0.19781050874891748, -‐1.4149733479708178, -‐1.4149733479708178, -‐1.4149733479708178, 0.5440680443502756, -‐1.4149733479708178]]
sequence = “GCATCGATGGCAGCGACTACAGCGCTACTACAGCGGAGACGATGCGATCGATACAAT”
hits = predictSites(sequence, PWM) print(hits) èè [3, 38, 43, 47]
Question 6 (20 points): Identification of putative target genesTranscription factors代写
Genes are portions of DNA sequences. The transcription start site of a gene is the position of the start of the gene in the sequence. Suppose that you have a dictionary of gene names and associated transcription start site position. For example
genes = {“BRCA1”:3, “MYC”:23, “RUNX”: 45}
Our goal is to count the number of predicted binding sites located in the neighborhood of the transcription start site of each gene. For example, if we allow a maximum distance of 10 and the list of PWM hits is [12, 38, 43, 46], then the BRCA1 gene has 1 hit, the MYC gene has 0, and the RUNX gene has 3.Transcription factors代写
Your task is to write the function countHitsPerGene, which takes as positional arguments the genes dictionary and the list of hits, as well as keyword argument maxDist, and returns a new dictionary with genes as keys and hit count as values.
Example of execution:
genes = {“BRCA1”:3, “MYC”:23, “RUNX”: 45} hits = [3, 38, 43, 47]
geneHits = countHitsPerGene(genes, hits, maxDist = 10) print(geneHits) èè {‘BRCA1’: 1, ‘MYC’: 0, ‘RUNX’: 3}
geneHits = countHitsPerGene(genes, hits, maxDist = 20) print(geneHits) èè {‘BRCA1’: 1, ‘MYC’: 3, ‘RUNX’: 3}

其他代写:考试助攻 计算机代写 java代写 function代写 paper代写 web代写 编程代写 report代写 数学代写 algorithm代写 金融经济统计代写 finance代写 python代写 java代写 code代写 代码代写 project代写 Exercise代写 作业帮助 app代写
